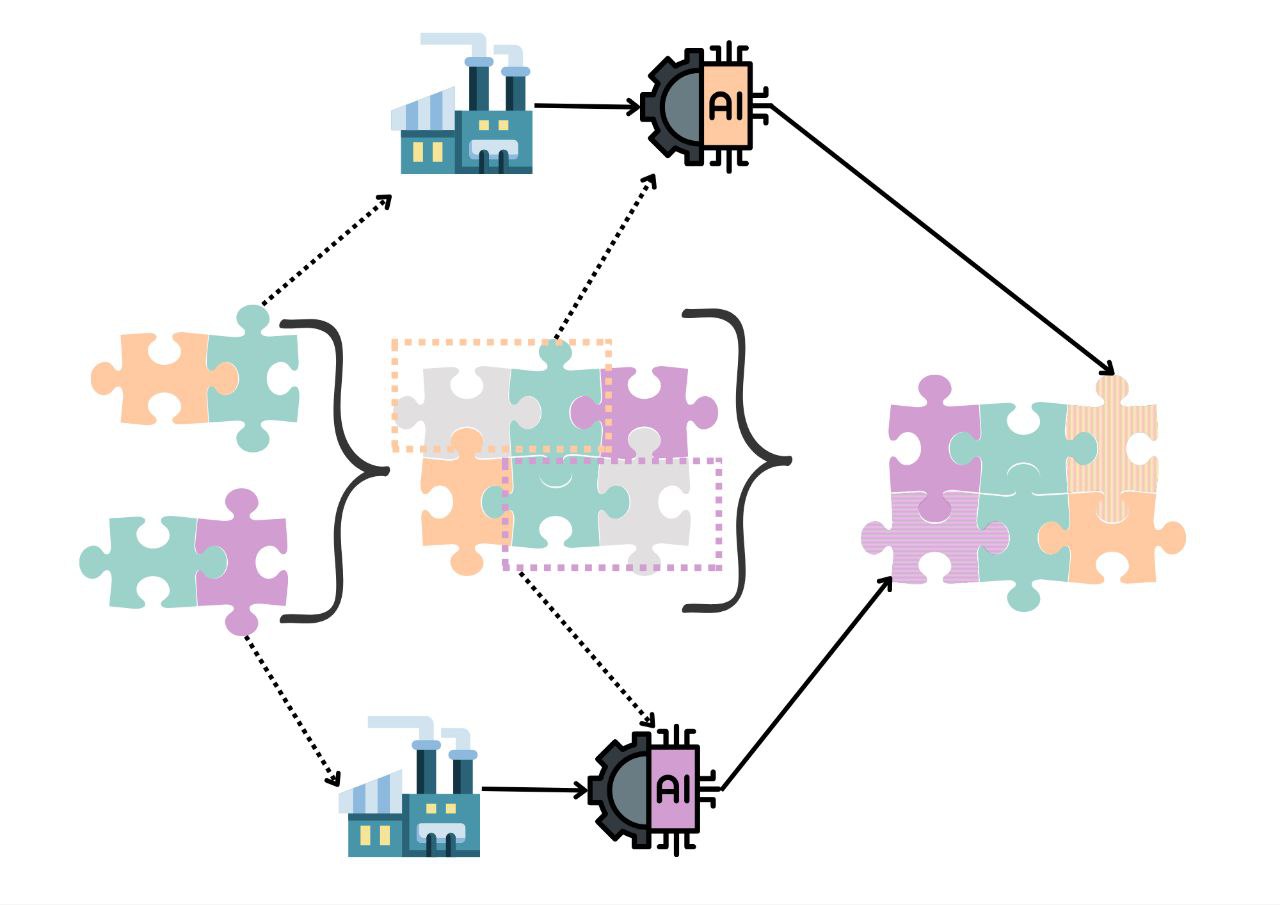
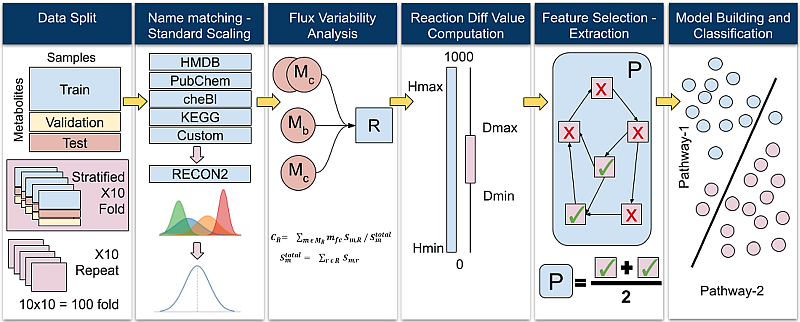
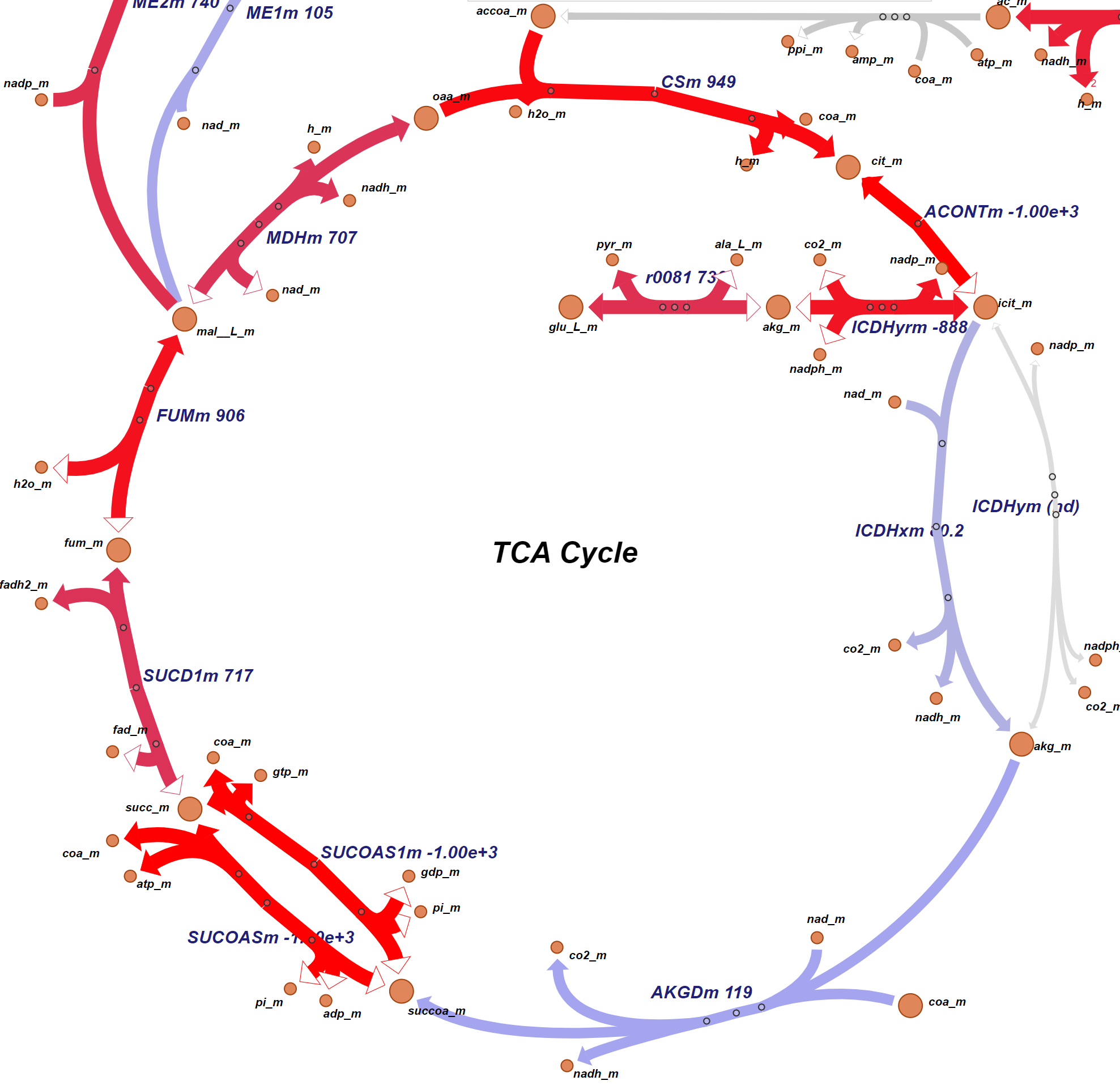
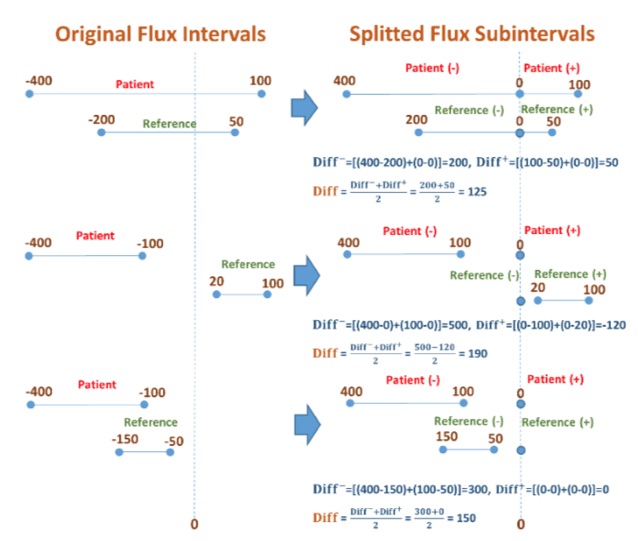
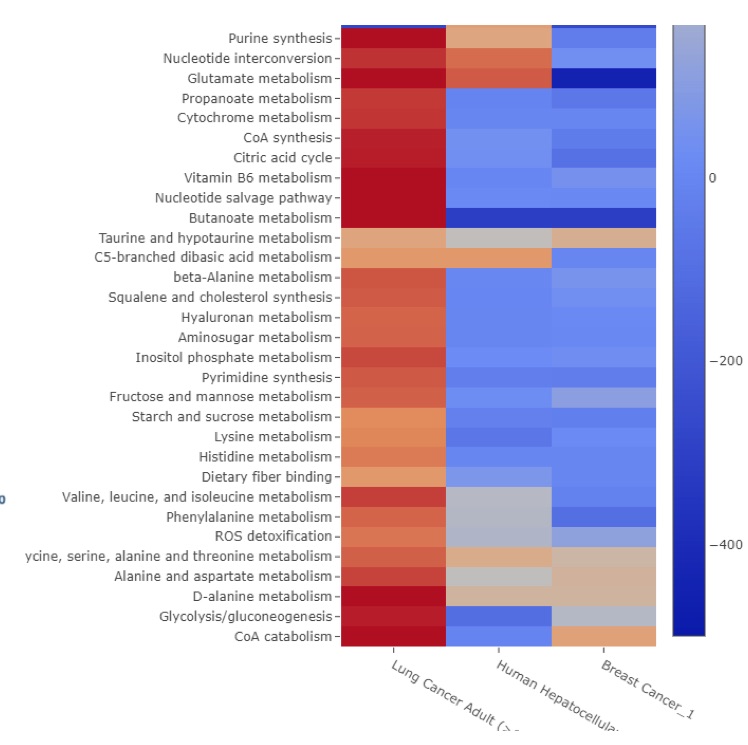
· Colorectal Cancer Risk Screening: This research explores the machine learning-based assessment of predisposition to colorectal cancer based on single nucleotide polymorphisms (SNP). Such a computational approach may be used as a risk indicator and an auxiliary diagnosis method that complements the traditional methods such as biopsy and CT scan. Moreover, it may be used to develop a low-cost screening test for the early detection of colorectal cancers to improve public health. We employ several supervised classification algorithms. We study SNPs in particular colorectal cancer-associated genomic loci that are located within DNA regions of 11 selected genes obtained from 115 individuals. We make the following observations: (i) random forest-based classifier using one-hot encoding and K-nearest neighbor (KNN)-based imputation performs the best among the studied classifiers with an F1 score of 89% and area under the curve (AUC) score of 0.96. (ii) One-hot encoding together with K-nearest neighbor-based data imputation increases the F1 scores by around 26% in comparison to the baseline approach which does not employ them. (iii) The proposed model outperforms a commonly employed state-of-the-art approach, ColonFlag, under all evaluated settings by up to 24% in terms of the AUC score. Based on the high accuracy of the constructed predictive models, the studied 11 genes may be considered a gene panel candidate for colon cancer risk screening (Cakmak et al. 2022).

Previous
Research:
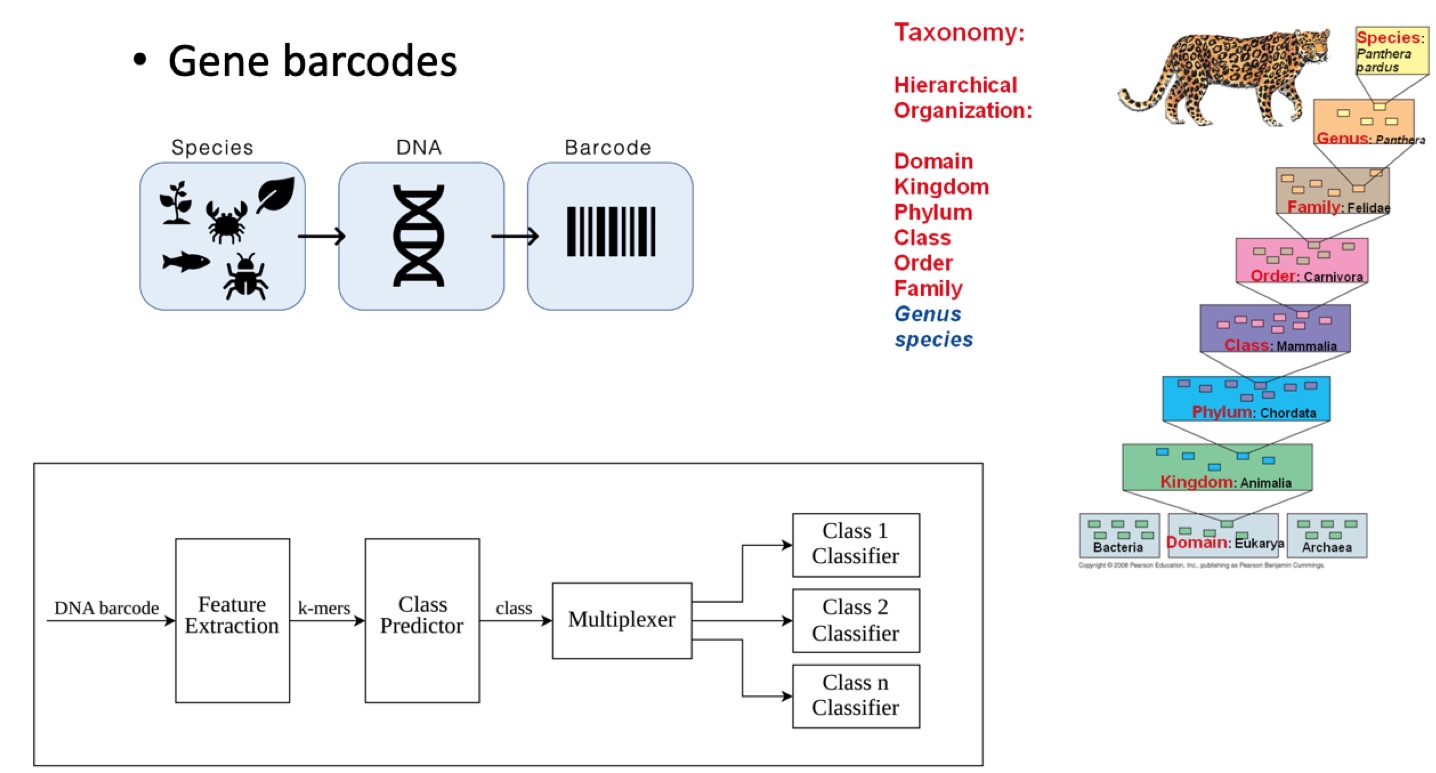
· Scalable Taxonomy Classification: Various
fields of applied biology (e.g., agriculture) depend on the classification of
living creatures. However, many popular and highly accurate machine learning
methods (e.g., Support Vector Machines) are not scalable to taxonomy settings
where there is a large number of labels/classes. In this research, we developed
a multi-level hierarchical classifier framework to automatically assign
taxonomy labels to DNA sequences (Sohsah et al., 2020). We utilize an
alignment-free approach called spectrum kernel method for feature extraction.
We demonstrate that the proposed framework provides higher accuracy (i.e., 95%)
than regular classifiers, and is scalable to taxonomy classification settings.
Furthermore, we show that the proposed framework is more robust to mutations
and noise in sequence data than the non-hierarchical classifiers.
· Query Selectivity Estimation: Query
optimizers of database management systems (DBMS) employ the expected size of a
query’s result set while automatically generating the most efficient execution
plan for the query. Therefore, accurately estimating the size of the query
result set (i.e., selectivity) is critical to the performance of DBMSs. In this
research, we first developed a new sequence pattern-based histogram structure
and an algorithm (i.e., SPH) (Aytimur and Cakmak, 2018) that employs the
patterns stored in this histogram to estimate the selectivity of fuzzy text
queries. SPH dramatically outperforms the state-of-the-art approaches for queries
with generic text patterns in terms of the estimation accuracy. Moreover, SPH
requires two orders of magnitude less space both in memory and on disk.
Besides, the selectivity estimation time of SPH is almost an order of magnitude
less in comparison to the state of the art. Next, in another work (Aytimur and
Cakmak, 2021), we developed novel positional sequence patterns. We demonstrated
that employing positional sequence patterns instead of regular sequence
patterns for fuzzy string selectivity estimation decreases the estimation error
by around 20%. Finally, we filed an international patent (Cakmak, 2018) that
describes the integration of the above-described techniques into a commercial
database management system.

· Digital Libraries: In today's academia,
publish or perish policy results in an enormous body of publications. Hence,
given the limited time, researchers have to be selective while putting a paper
into their reading list, as well as prioritizing the articles in that list.
Ideally, many would be more interested in reading papers that are likely to
have a high impact on their fields. However, it is almost impossible to decide
whether a paper would really make a high impact ahead of time before reading it
(and often even after reading it). In this research (Davletov et al., 2014), we
developed an impact prediction framework for academic papers. Our model uses a time-series
approach to predict the number of citations. In particular, our model makes use
of citation behavior, i.e., the pattern of increase in the number of citations.
In the training phase, the papers are clustered according to citation
behaviors. Then, when a new paper is published, it is assigned to a cluster and
the prediction is performed accordingly.
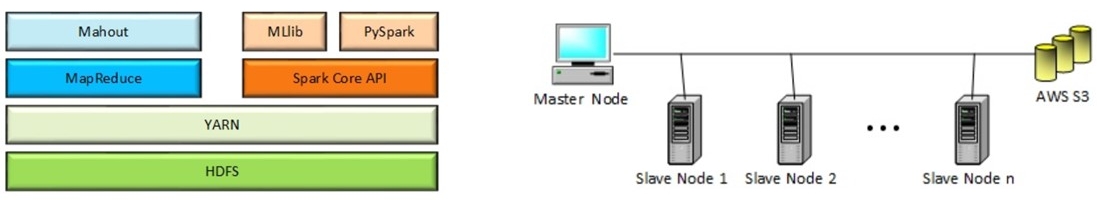
· Machine Learning on Big Data: Most of the popular Big Data analytics tools evolved to adapt their
working environment to extract valuable information from a vast
amount of unstructured data. The ability of data mining techniques
to filter this helpful information from Big Data led to the term ‘Big
Data Mining’. Shifting the scope of data from small-size, structured,
and stable data to huge volume, unstructured, and quickly changing
data brings many data management challenges. Different tools cope
with these challenges in their own way due to their architectural
limitations. There are numerous parameters to take into consideration when choosing the right data management framework based
on the task at hand. In this paper, we present a comprehensive
benchmark for two widely used Big Data analytics tools, namely
Apache Spark and Hadoop MapReduce, on a common data mining
task, i.e., classification. We employ several evaluation metrics to
compare the performance of the benchmarked frameworks, such
as execution time, accuracy, and scalability. These metrics are specialized to measure the performance for classification task. To the
best of our knowledge, there is no previous study in the literature that employs all these metrics while taking into consideration
task-specific concerns. We show that Spark is 5 times faster than
MapReduce on training the model. Nevertheless, the performance
of Spark degrades when the input workload gets larger. Scaling the
environment by additional clusters significantly improves the performance of Spark. However, similar enhancement is not observed
in Hadoop. Machine learning utility of MapReduce tend to have
better accuracy scores than that of Spark, like around 2%-3%, even
in small-size data sets. (Tekdogan and Cakmak, 2021).
· Mining for Unknown Pathways: Many
essential biological pathways still remain unknown or incomplete for newly
sequenced organisms. Moreover, experimental validation of enormous numbers of
possible pathway candidates in a wet-lab environment is time and
effort-extensive. In this research, we developed comparative genomics tools and
algorithms (Cakmak and Ozsoyoglu, 2007a; Cakmak et al., 2007; Ratprasartporn et
al., 2006; Cakmak and Ozsoyoglu, 2008a) that help scientists predict pathways
in an organism’s metabolic network (with 86% precision and 74% recall).
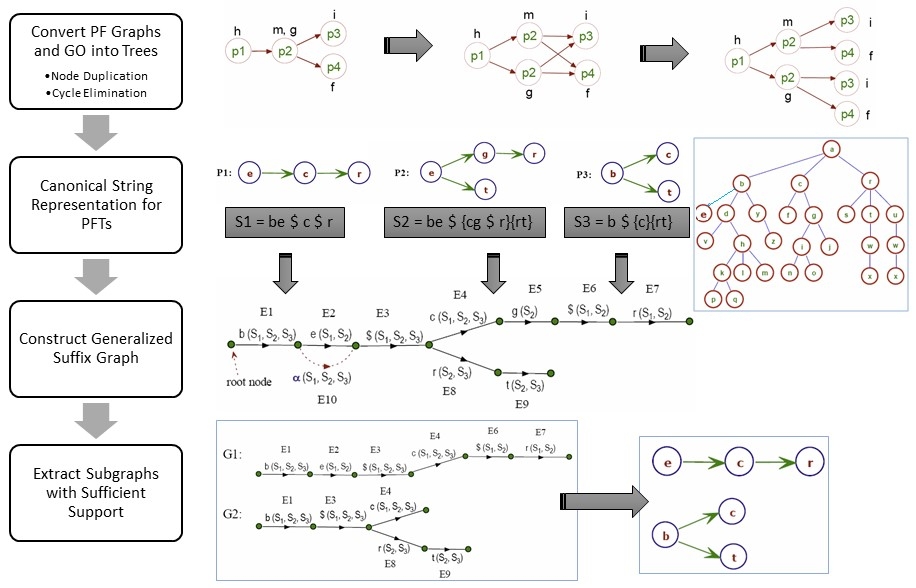
· Automatic Inference of Gene/Protein Annotations
from Literature through Text Mining: Genes and proteins are
frequently annotated with the Gene Ontology (GO) concepts. The most reliable GO
annotations of genes and gene products are created by biologists manually
reading related papers and determining the proper GO concepts to be assigned to
the corresponding genes. Nevertheless, locating and curating information about
a genomic entity from the biomedical literature requires vast amounts of human
effort. In this research, we developed automated text mining tools (Cakmak and
Ozsoyoglu, 2008b; Cakmak and Ozsoyoglu, 2007b; Ratprasartporn et al., 2009) to
annotate genes and gene products with the Gene Ontology concepts via capturing
the related knowledge embedded in textual data to expedite and automate the
annotation of genomic entities by GO concepts. The proposed algorithm has
reached 78% precision and 61% recall.

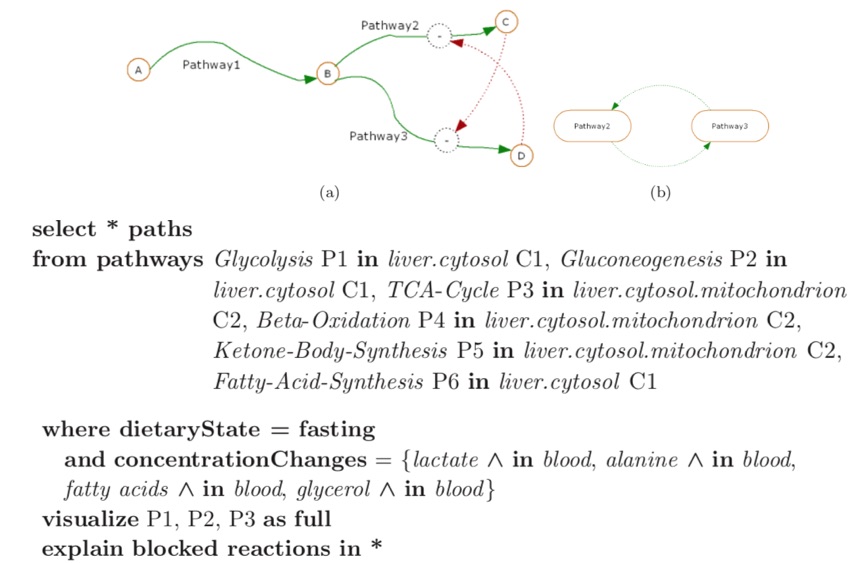
· Metabolism Query Language: In another
research, we designed a “metabolism query language” (MQL) (Cakmak et al., 2010;
Cakmak et al., 2012; Cicek et al. 2014) that computationally captures the
metabolism data and allows to query it at a detailed level. MQL accommodates
metabolic network knowledge in a manner faithful to the underlying
biochemistry. We also solve and present query processing techniques for MQL
queries.
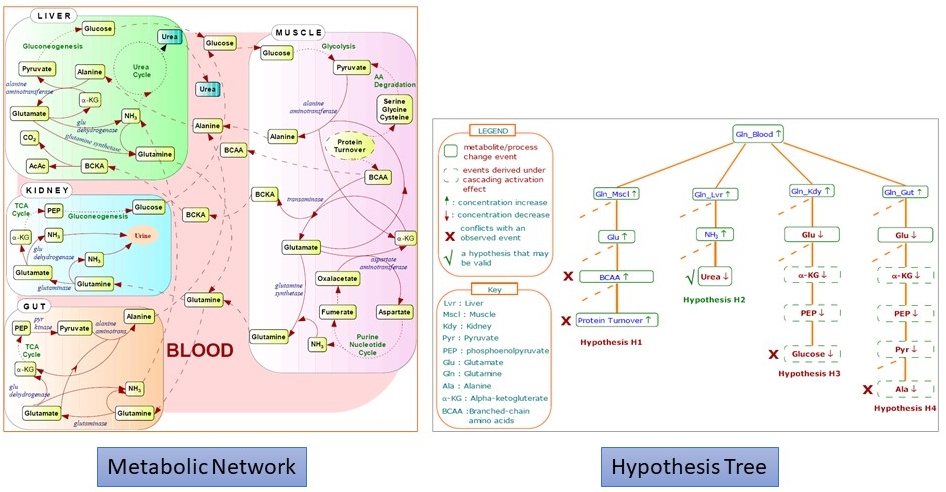
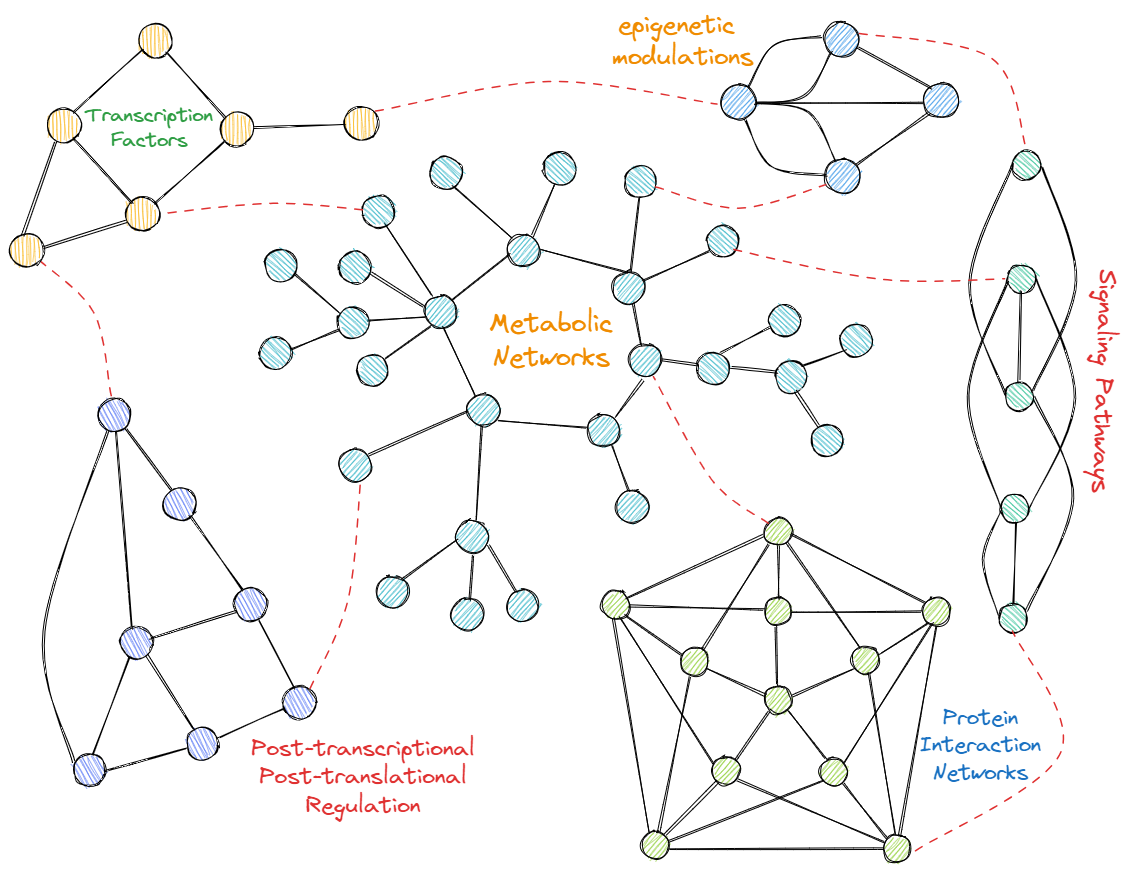
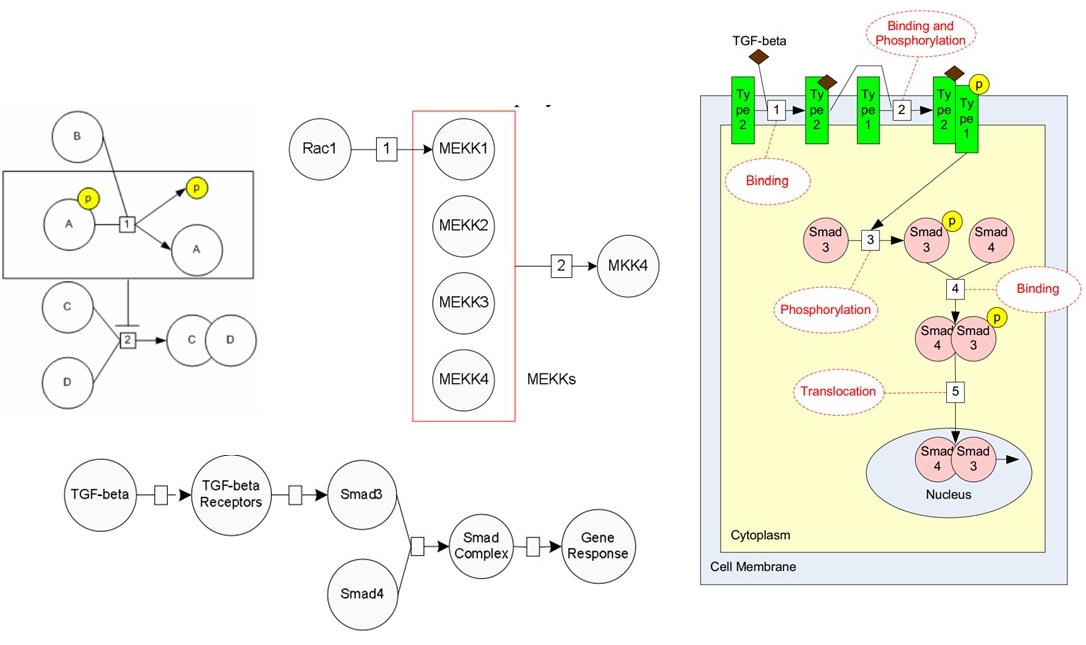
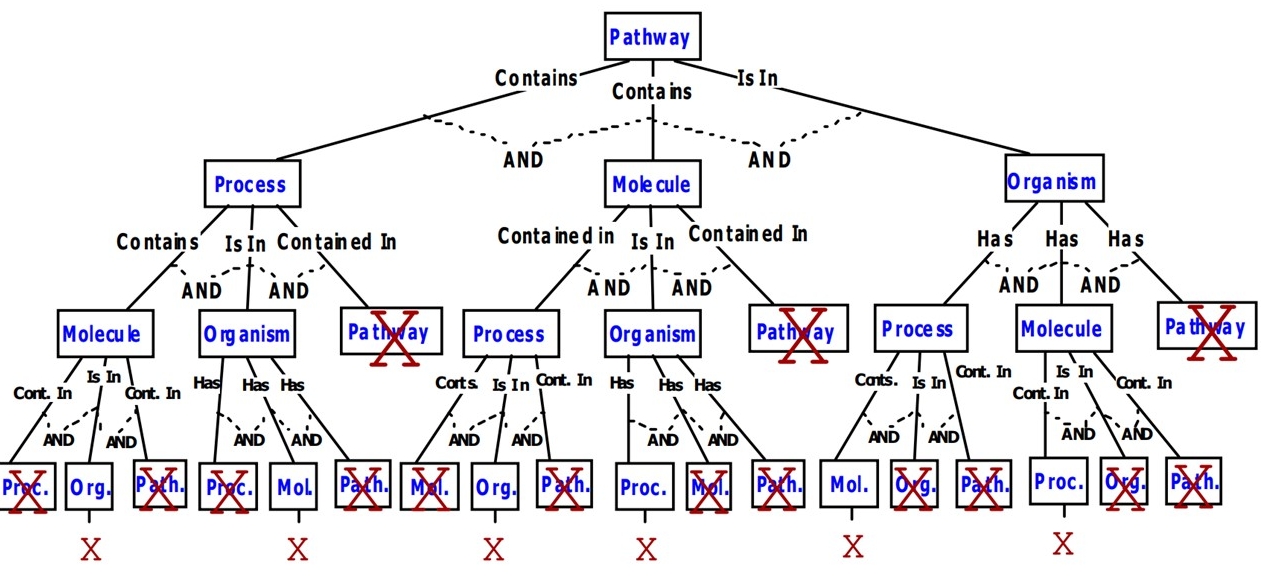
· Data & Visualization Models for Pathways: Signaling pathways are chains of interacting proteins,
through which the cell converts a (usually) extracellular
signal into a biological response. The number of known
signaling pathways in the biological literature and on the
web has been increasing at a very high rate, thus
demanding a need for efficient ways of storing, visualizing,
querying, and mining signaling pathways. In this work,
first we briefly compare the data modeling and
visualization capabilities of existing signaling pathways
systems. Then, we present a signaling pathway data model
and its visualization that subsumes the existing models.
Our model visualizes a signaling pathway (a) as a
nested graph, (b) with explicit location information (e.g.,
cell, tissue, organelle, nucleus, etc.), and (c) in four
abstraction levels, namely, the levels of molecule-tomolecule signaling steps, collapsed sub-pathways,
molecule-to-pathway connections, and pathway-to-pathway
connections. We model (1) the effects of specific signaling
steps, (2) state changes of signaling molecules, (3) various
(extensible) structural/physical changes of signaling
molecules such as complex formation, dissociation,
assembly, oligomerization, di-/trimerization, cleavage and
degradation, (4) condensation/hydrolysis signaling steps,
and (5) exchanges and translocations as signaling steps.
The visualization model gracefully models incomplete
information and hierarchical levels of signaling molecules.
Finally, we introduce a completely new visualization
dimension for pathways, namely, Gene Ontology (GO)-
based functional visualizations of pathways. We believe
that functional visualizations of pathways provides new
opportunities in understanding, defining and comparing
existing pathways, and in helping discover new ones.
· Biological Web Data Source Integration: Biological web data sources have now become essential
information sources for researchers. However, their use is tedious,
labor-intensive, repetitive, and possibly involve the integration of
data from multiple web data sources. In this paper, as a first step
towards the full integration of web data sources, we propose a
framework that allows an integrated use of biological sources in a
task-oriented manner. We define and experimentally evaluate a
toolkit-based framework for semi-automatically constructing an
integrated (software) system that automates and optimizes the
execution of a biology-related computational task at hand. To test
and refine the principles of the framework, we build and evaluate
“Pathway-Infer” as a benchmark integrated system.
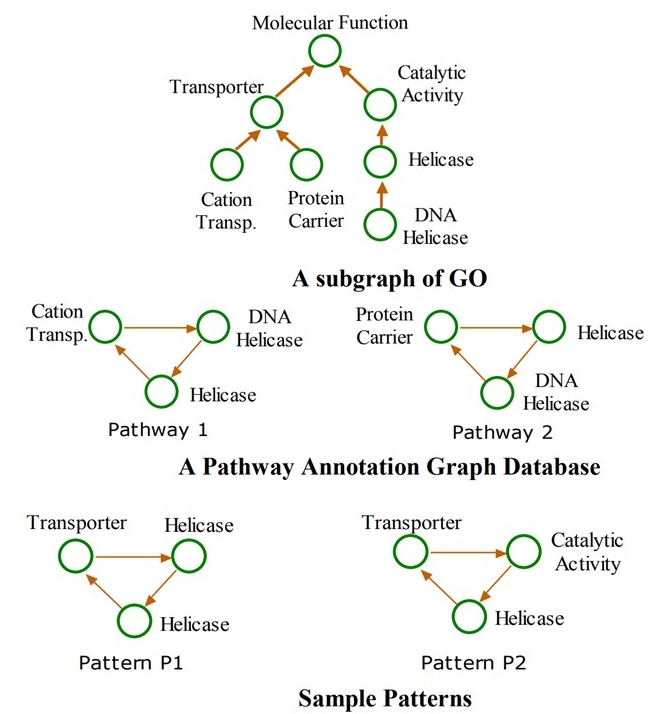
· Taxonomy-superimposed Graph Mining: New graph structures where node labels are members of
hierarchically organized ontologies or taxonomies have become
commonplace in different domains, e.g., life sciences. It is a
challenging task to mine for frequent patterns in this new graph
model which we call taxonomy-superimposed graphs, as there may
be many patterns that are implied by the
generalization/specialization hierarchy of the associated node label
taxonomy. Hence, standard graph mining techniques are not
directly applicable.
In this work, we develop Taxogram, a taxonomy-superimposed
graph mining algorithm that can efficiently discover frequent
graph structures in a database of taxonomy-superimposed graphs.
Taxogram has two advantages: (i) It performs a subgraph
isomorphism test once per class of patterns which are structurally
isomorphic, but have different labels, and (ii) it reconciles
standard graph mining methods with taxonomy-based graph
mining and takes advantage of well-studied methods in the
literature. Taxogram has three stages: (a) relabeling nodes in the
input database, (b) mining pattern classes/families and
constructing associated occurrence indices, and (c) computing
patterns and eliminating useless (i.e., over-generalized) patterns
by post-processing occurrence indices. Experimental results show
that Taxogram is significantly more efficient and more scalable
compared to other alternative approaches.
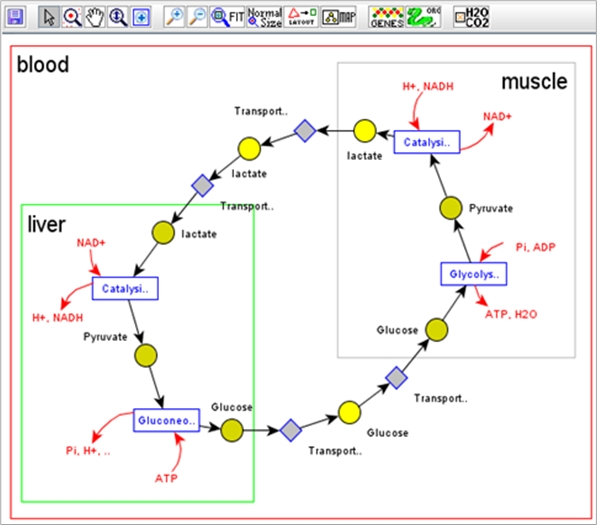
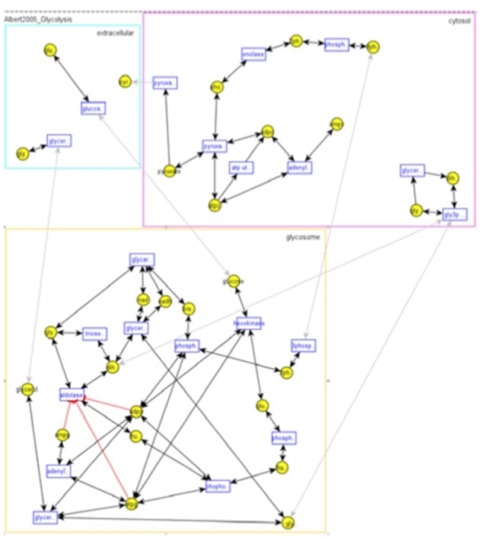
· Systems Biology Work: In this
research, we designed and developed a Systems Biology platform (Cakmak et al.,
2011; Coskun et al., 2012) that brings together, under a single database
environment, metabolic pathways data and systems biology models. Besides, it
provides expanded browsing, querying, visualization, and simulation
capabilities to help with systems biology modeling and analysis, all brought
about due to the integrated environment. This platform is built upon a pathways
database system that we developed earlier (Elliott, 2008).
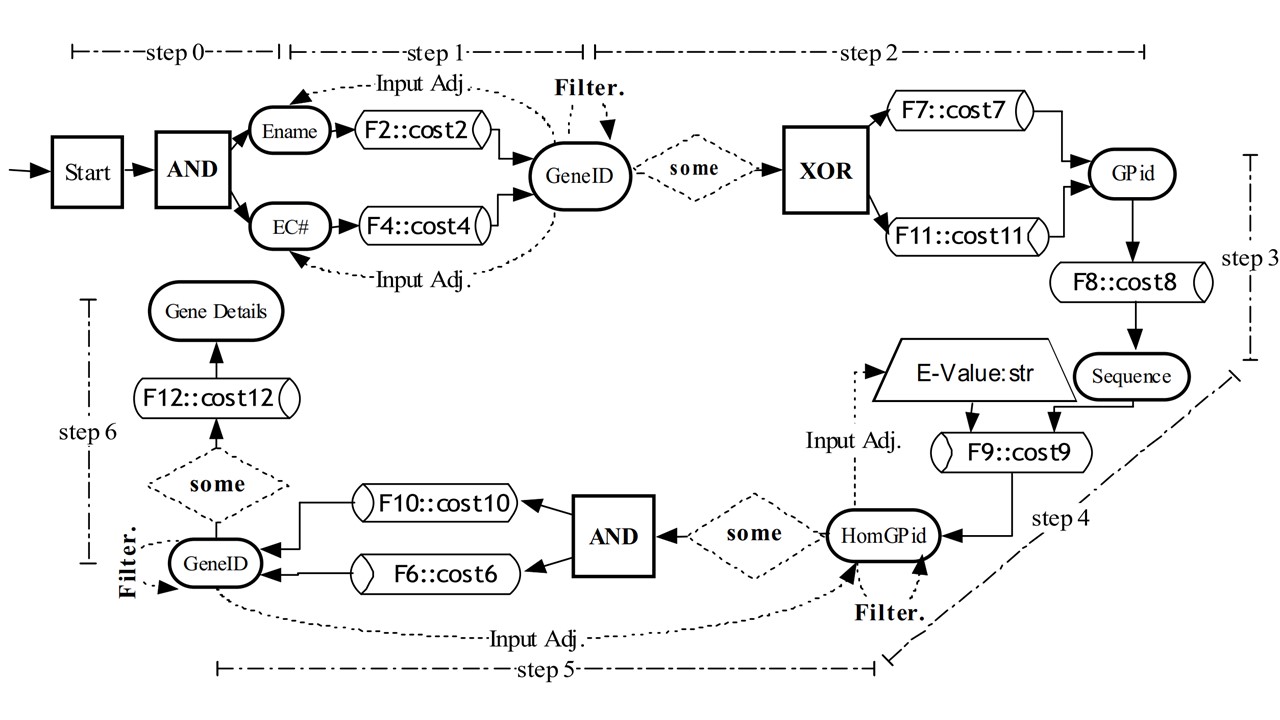
· Advanced Querying for Biological Networks: Querying biochemical networks in flexible ways over the web is
important to facilitate ongoing biological research. In this work,
we present a querying interface for biological networks, more
specifically, metabolic networks. The interface allows for the
specification of a large class of containment, path, and
neighborhood queries with ease from a web browser. The query
specification process is user-friendly, employs hierarchically
arranged relationships among biological entities, and uses autocomplete features. The interface is provided as part of PathCase, a
system to store, query, visualize and analyze metabolic pathways
at different levels of detail.
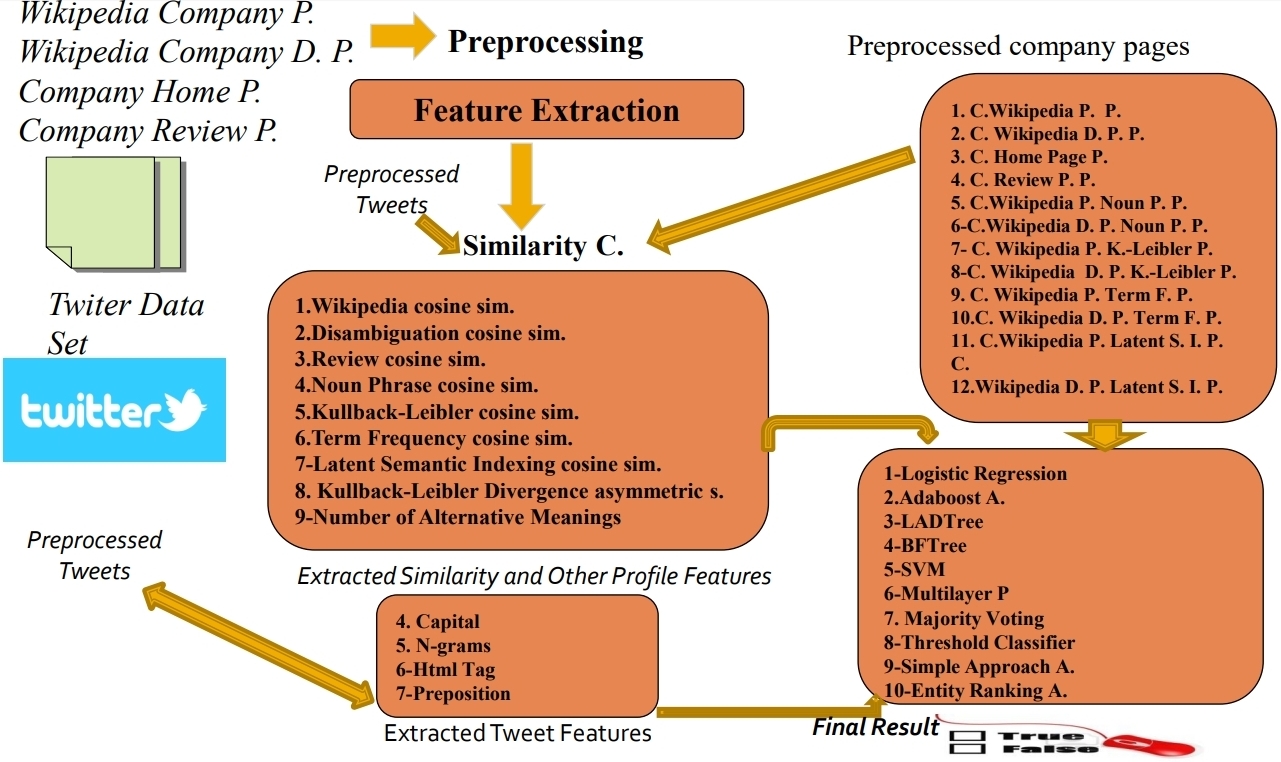
· Mining Twitter for Company Mentions: Twitter is an online social networking website where people can post short messages on any subject, and
these messages become visible to other users. Users intentionally express their opinions about companies or products
via microblogging texts. Analyzing such messages might help explore what customers think about company products,
or what the broad feelings of customers are. Identifying tweets referring to products and companies is becoming an
important tool recently. However, company names are often vague. Hence, the first step is to locate the messages that
are relevant to a company. In this paper, we present a number of supervised learning techniques to decide whether a given
tweet is about a company, e.g., whether a message containing the term ‘amazon’is related to the company Amazon Inc.
or not. Solving this task is challenging in comparison to the classical classification process. The main difficulty with this
problem is that tweets and company names include limited information. To make this task tractable, external resources
are used to get richer data about a company. More specifically, we generate several profiles for each organization, which
contain richer information. Then we perform feature extraction to obtain both numerical and categorical features and we
do feature selection to identify the most relevant attributes with our task. Finally, we train several supervised classifiers.
Our constructed classifiers and carefully selected features provide high accuracy on the WePS-3 dataset.
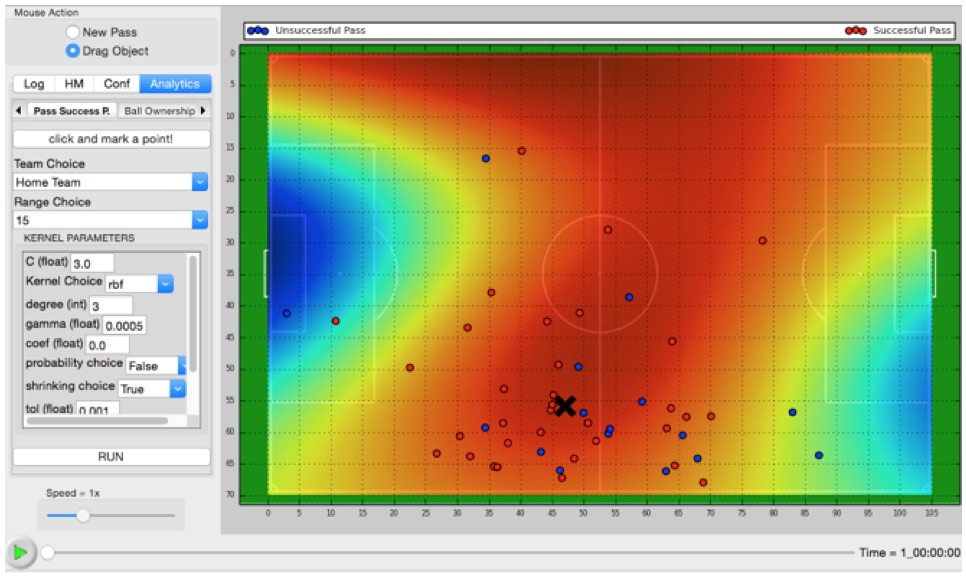
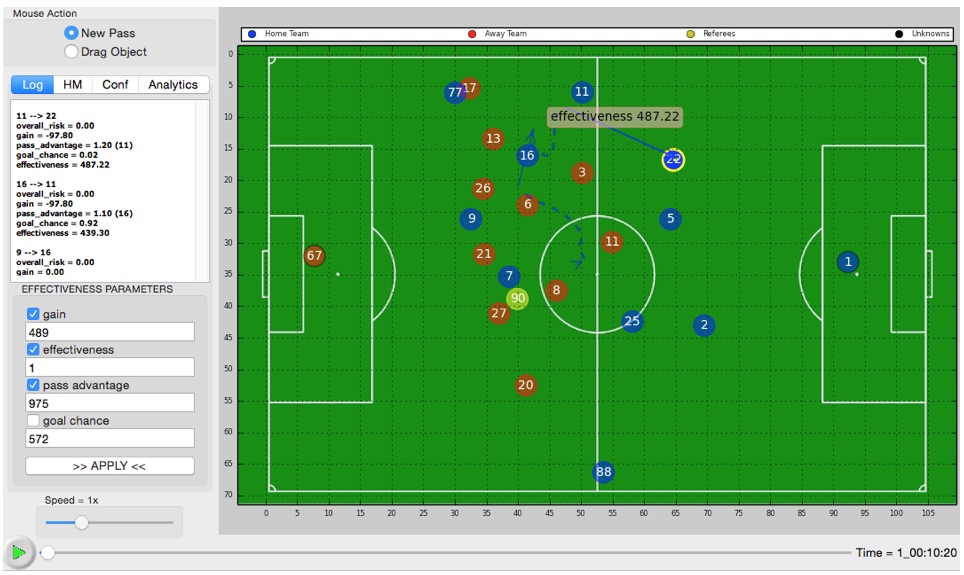
· Sports Analytics: In recent years,
computerized tracking systems that can collect spatiotemporal data from soccer
games have become commonplace in major leagues and international competitions.
In this research, we developed an interactive visual analytics tool (Delibas et
al., 2019a) that can be used to perform an exploratory analysis of soccer data.
The visualizations are enriched by utilizing various data mining and machine
learning algorithms in the backend. The tool also provides advanced analytics
options such as pass success, ball ownership, optimal shooting point, and pass
effectiveness prediction (Cakmak et al., 2018). We also filed an international
patent (Delibas et al., 2019b) that describes an intelligent system to help
players make the most optimal decision during training sessions by taking
advantage of the aid of a wearable device similar to the Google Glass with
instant feedback.
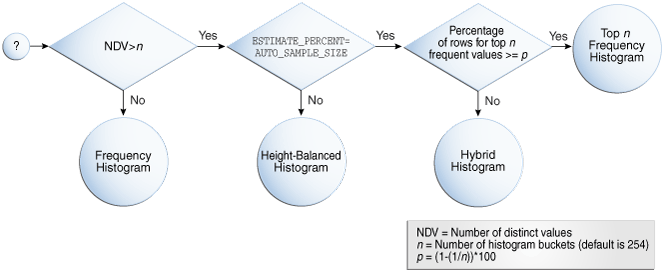
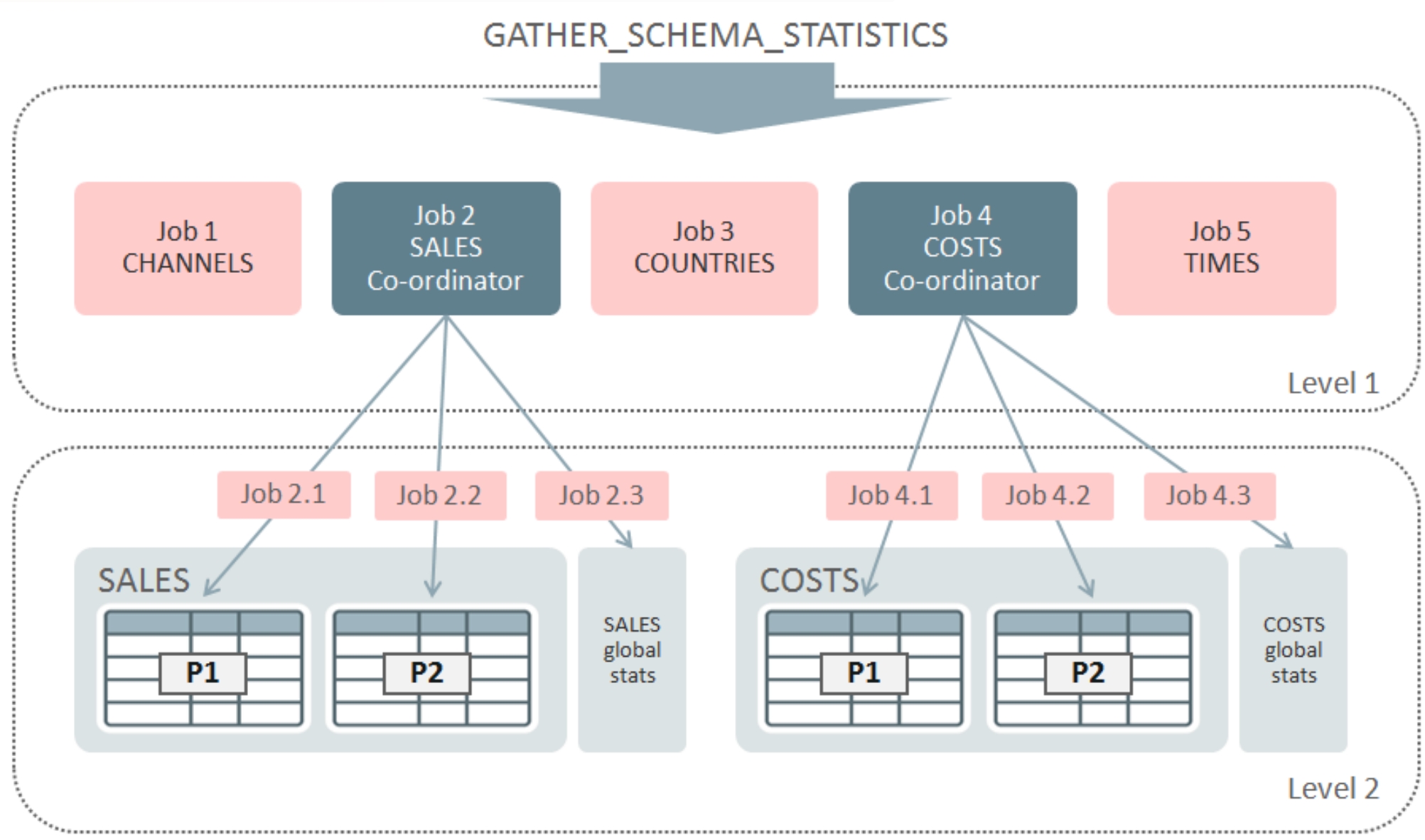
· Query Optimization Work: During my 4
years tenure at Oracle, Inc. (San Francisco, CA), I developed two novel
histogram structures, namely, Hybrid Histograms and Frequency-based Histograms,
which provide more accurate predicate selectivity estimations during query
optimization. Furthermore, I also developed a framework that gathers database
statistics for multiple objects in a highly scalable manner that improved
statistics gathering time up to 10x. These developments are included in two
international patent filings (Chakkappen et al., 2016; Chakkappen et al.,
2017). Furthermore, these features are released to the users in Oracle’s market
leader Database Management System flagship product in version 12c (Belknap et
al., 2013).
-
Bibliography:
Aytimur, M., & Cakmak,
A. (2018). Estimating the selectivity of LIKE queries using pattern-based
histograms. Turkish Journal of Electrical Engineering & Computer Sciences,
26(6), 3319-3334.
Aytimur, M., Cakmak, A. (2021). Using positional sequence patterns
to estimate the selectivity of SQL LIKE queries. Expert Systems with Applications, 165, 113762.
Belknap, P., Cakmak, A., Chakkappen, S., Chan, I.,
Chatterjee, D., Das, D., ... & Lee, A. (2013). Oracle Database SQL Tuning
Guide, 12c Release 1 (12.1) E15858-15.
Cakmak, A. LIKE
Selectivity Estimation. (2018) International Patent App. PCT/TR2018/050366.
Cakmak, A., Qi, X.,
Coskun, S. A., Das, M., Cheng, E., Cicek, A. E., Lai, N., Ozsoyoglu, G. &
Ozsoyoglu, Z. M. (2011). PathCase-SB architecture and database design. BMC
systems biology, 5(1), 188.
Cakmak, A., Qi, X.,
Cicek, A. E., Bederman, I., Henderson, L., Drumm, M., & Ozsoyoglu, G.
(2012). A new metabolomics analysis technique: steady-state metabolic network
dynamics analysis. Journal of bioinformatics and computational biology, 10(01),
1240003.
Cakmak, A., Celik, MH.
(2018). A System for Diagnosing Diseases. International Patent App. PCT/TR2019/051101.
Cakmak, A., Kirac, M.,
Reynolds, M. R., Ozsoyoglu, Z. M., & Ozsoyoglu, G. (2007, July). Gene
ontology-based annotation analysis and categorization of metabolic pathways. In
19th International Conference on Scientific and Statistical Database Management
(SSDBM 2007) (pp. 33-33). IEEE.
Cakmak, A., &
Ozsoyoglu, G. (2007a). Mining biological networks for unknown pathways.
Bioinformatics, 23(20), 2775-2783.
Cakmak, A., &
Özsoyoglu, G. (2007b). Annotating genes by mining PubMed. In Pacific Symposium
on Biocomputing.
Cakmak, A., &
Ozsoyoglu, G. (2008a). Taxonomy-superimposed graph mining. In Proceedings of
the 11th international conference on Extending database technology: Advances in
database technology (pp. 217-228).
Cakmak, A., &
Ozsoyoglu, G. (2008b). Discovering gene annotations in biomedical text
databases. BMC bioinformatics, 9(1), 143.
Cakmak, A., Ozsoyoglu,
G., & Hanson, R. W. (2010). Querying metabolism under different
physiological constraints. Journal of bioinformatics and computational biology,
8(02), 247-293.
Cakmak, A., Uzun, A.,
& Delibas, E. (2018). Computational modeling of pass effectiveness in
soccer. Advances in Complex Systems, 21(03n04), 1850010.
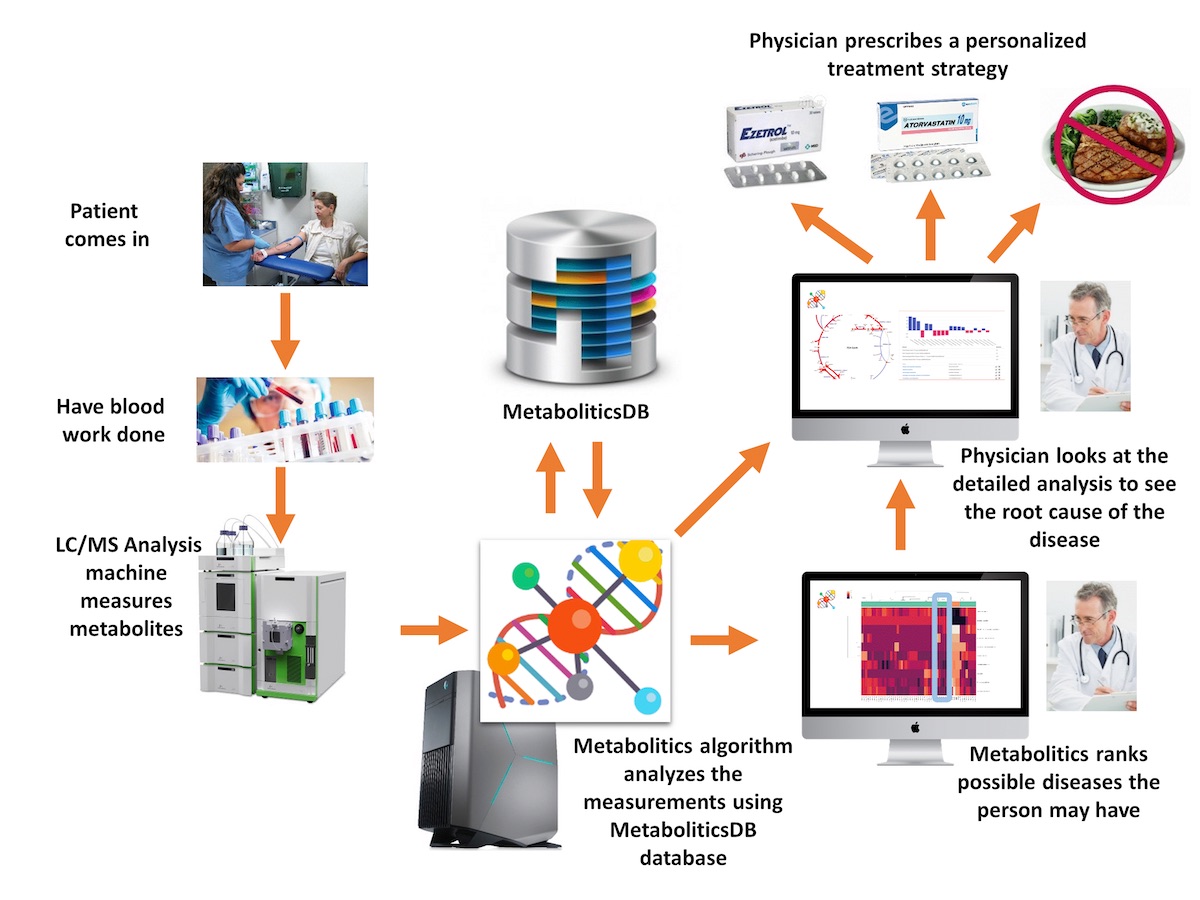
Cakmak, A., &
Celik, M. H. (2020). Personalized Metabolic Analysis of Diseases. IEEE/ACM Transactions
on Computational Biology and Bioinformatics (in press).
Celik, M. H., Saleh,
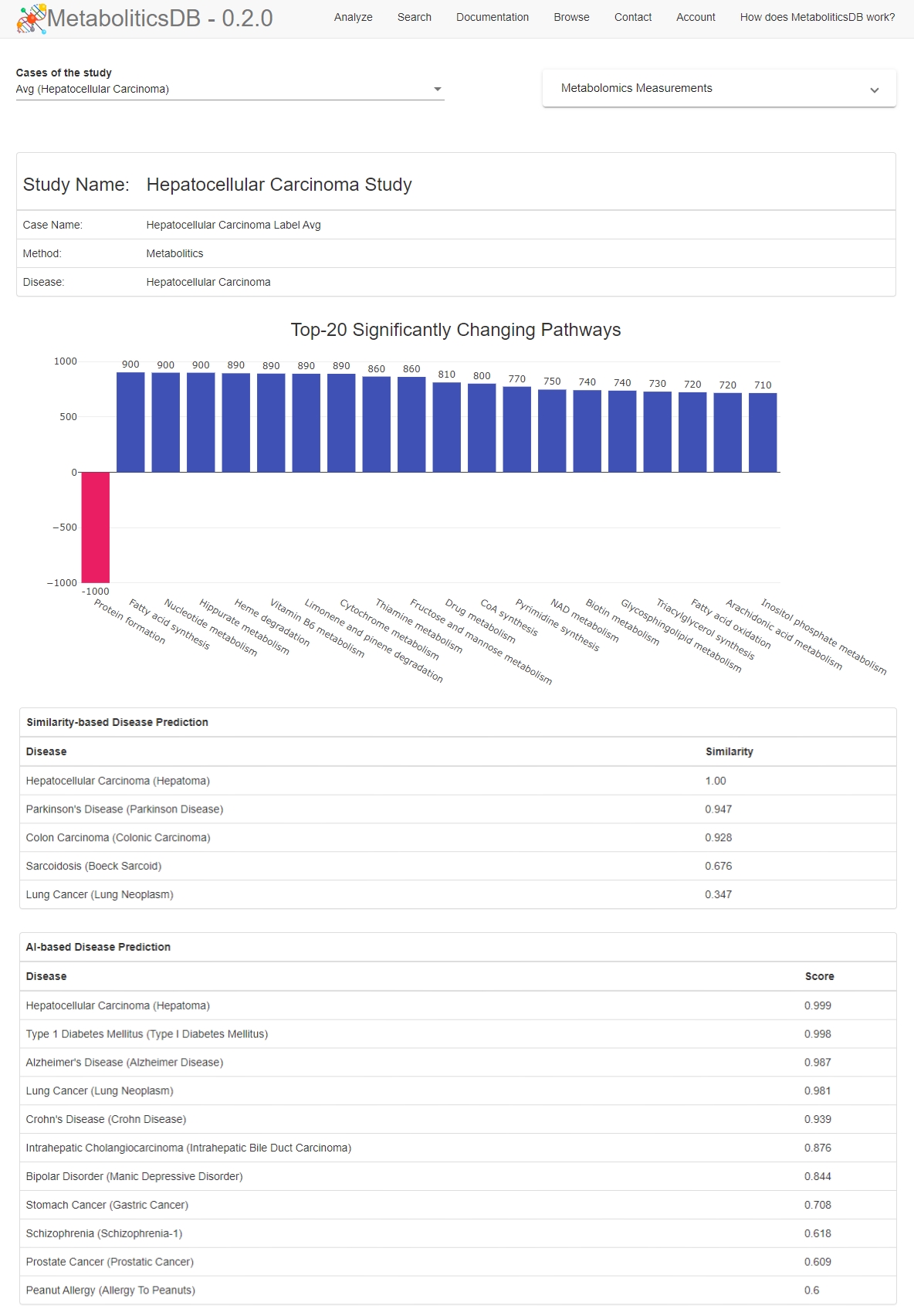
T., Dokay, A., Cakmak, A. (2020). MetaboliticsDB: a
database of Metabolomics analyses. (Under major revision for IEEE/ACM
Transactions on Computational Biology and Bioinformatics). The web tool is
available online at http://metabolitics.biodb.sehir.edu.tr/
.
Cicek, A.E., Qi, X.,
Cakmak, A., Johnson, S.R., Han, X., Alshalwi, S., Ozsoyoglu, Z.M. and Ozsoyoglu,
G., 2014. An online system for metabolic network analysis. Database (Oxford
Univ. press).
Chakkappen, S. P.,
Zait, M., Lee, A. W., & Cakmak, A. (2016). U.S. Patent No. 9,471,631.
Washington, DC: U.S. Patent and Trademark Office.
Chakkappen, S. P.,
Zait, M., Lee, A. W., & Cakmak, A. (2017). U.S. Patent Application No.
15/295,539.
Coskun, S.A., Qi, X.,
Cakmak, A., Cheng, E., Cicek, A.E., Yang, L., Jadeja,
R., Dash, R.K., Lai, N., Ozsoyoglu, G. and Ozsoyoglu, Z.M. (2012). PathCase-SB:
integrating data sources and providing tools for systems biology research. BMC
systems biology, 6(1), p.67.
Davletov, F., Aydin, A.
S., & Cakmak, A. (2014, November). High impact academic paper prediction
using temporal and topological features. In Proceedings of the 23rd ACM
International Conference on Conference on Information and Knowledge Management
(pp. 491-498).
Delibas, E., Uzun, A.,
Inan, M. F., Guzey, O., & Cakmak, A. (2019a). Interactive exploratory
soccer data analytics. INFOR: Information Systems and Operational Research,
57(2), 141-164.
Delibas, E., Uzun, A.,
Inan, M. F., Guzey, O., & Cakmak, A. (2019b). U.S. Patent Application No.
16/331,744.
Elliott, B., Kirac, M.,
Cakmak, A., Yavas, G., Mayes, S., Cheng, E., ... & Meral Ozsoyoglu, Z.
(2008). PathCase: pathways database system. Bioinformatics, 24(21), 2526-2533.
Ratprasartporn, N.,
Cakmak, A., & Ozsoyoglu, G. (2006, July). On data and visualization models
for signaling pathways. In 18th International Conference on Scientific and
Statistical Database Management (SSDBM'06) (pp. 133-142). IEEE.
Ratprasartporn, N., Po,
J., Cakmak, A., Bani-Ahmad, S., & Ozsoyoglu, G. (2009). Context-based
literature digital collection search. The VLDB Journal, 18(1), 277-301.
Sohsah, G. N., Ibrahimzada, A. R., Ayaz, H., Cakmak, A. (2020).
Scalable classification of organisms into a taxonomy using hierarchical supervised learners. Journal of Bioinformatics and Computational Biology, 18(05), 2050026.